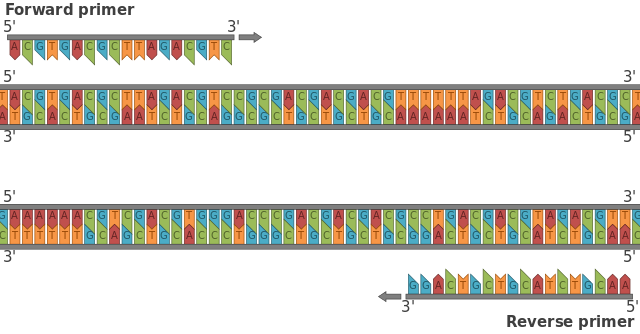
¿Qué es un primer?
Los primers son secuencias cortas de moléculas de ácidos nucléicos (entre 18 a 24 pares de bases) que son utilizados para la amplificación de un gen o un fragmento de ADN de interés, mediante la Reacción en Cadena de la Polimerasa (PCR).
De tal forma que los cebadores o primers, son uno de los principales ingredientes para una reacción de PCR, y de ellos depende la especificidad, porque al unirse complementariamente a las dos cadenas de ADN de la secuencia molde, fijan por así decirlo las coordenadas donde se llevará acabo la reacción.
Para poder obtener un par de primers eficientes, se necesita un análisis cuidadoso de la región de interés porque hay muchos factores, que pueden influir, por ejemplo, los primers podrían ser complementarios entre ellos o formar estructuras secundarias, lo que podría resultar en obtener amplificaciones de fragmentos no deseados fuera del blanco de la región de interés.
Sin embargo, gracias al diseño asistido por computadora, es relativamente fácil poder obtener un par de buenos primers.
Podemos resumir en las siguientes reglas, los pasos a seguir para poder buscar buenos candidatos para los cebadores.
Reglas para el diseño de los primers
- Carácter único: Asegura que solo exista un sitio de unión al ADN molde
- Tamaño: de 18 a 24 pares de base
- Composición de bases: %GC de 50-60 evita regiones ricas en (A+T) y (C+G)
- Evitar tener más de 3 bases GC en los extremos del primer
- Tm se prefieren valores entre 55 y 80 C
- Asegurarse que no existe una diferencia de Tm mayor de 2 o 3 unidades entre ellos
- Minimizar el efecto de las estructuras secundarias.
- Valores aceptables de $\Delta G$
- Evitar valores menores a: -9 $\frac{Kcal}{mol}$
- Harpins -2 $\frac{Kcal}{mol}$
- Dímeros internos -6 $\frac{Kcal}{mol}$
- Complementariedad Auto/cruzada extremo 3’-5 $\frac{Kcal}{mol}$
- Valores aceptables de $\Delta G$
Además de considerar estas reglas Aoyagi (2001, pp 312 - 313) propone algunas preguntas para definir nuestros objetivos para el diseño de los primers
Objetivos para el diseño de los primers
- ¿Qué debe llevar a cabo la PCR?
- Que presión pone esto en los primers
- ¿Se deben identificar uno o varios blancos?
- La identificación de varios blancos requiere de un numero de primers que puede incrementar la dificultad de evitar el sobrelapamiento en el extremo 3'
- ¿Se debe clonar la región completa de una región codificante?
- Para un producto largo se debe utilizar el algoritmo nearest-neighbor para la selección de una $\mathrm{T}_{m}$ adecuada.
- ¿Se deben generar datos cuantitativos?
- En este caso la eficiencia de la PCR es crítica, así que deben de evitarse en la medida de lo posible la formación de dímeros.
- ¿Se debe amplificar una secuencia (homóloga) basándose en la información de otra especie?
- En este caso debe partirse de el alineamiento de muchas secuencias para poder seleccionar la región más conservada para que el diseño del primer incremente la probabilidad de éxito.
- ¿Se debe evitar la amplificación de pseudogenes?
- ¿Qué se conoce acerca de los pseudogenes que corresponden a la diana de la secuencia de interes?
- Una revisión preliminar de literatura puede ahorrar muchos dolores de cabeza, porque existen muchos pseudogenes reportados.
- Un forma fácil para buscar la amplificación de pseudogenes es por medio de una búsqueda en BLAST
- La única forma de evitar los pseudogenes es el diseño de primers através de las uniones exón-exón y probarlos en el laboratorio amplificando el DNA genómico.
- Los pseudogenes procesados no poseen intrones, de tal forma que pueden ser amplificados en la extensión de los primers de PCR sobre las dos uniones de exones.
- ¿Qué se conoce acerca de los pseudogenes que corresponden a la diana de la secuencia de interes?
- ¿Se están buscando datos acerca de polimorfismo de nucleótidos (SNP)?
- En este caso se requiere de estrategias especializadas.
- ¿Debe de diseñarse un amplicón pequeño para incrementar la detección del gen en muestras donde sería muy dificil amplificar un fragmento largo?
- Por ejemplo, muestras forenses, secciones embebidas de parafina o parcialmente degradadas.
Metas para el diseño
A que nos referimos con Especificidad y Eficiencia de amplifiación
- Especificidad: Frecuencia de eventos propios de hibridación con el ADN molde (priming), si el primer no es específico se pueden amplificar secuencias fuera del blanco (mis-priming).
- Eficiencia: Incremento de producto en cada ciclo de PCR
En las aplicaciones diagnósticas se puede sacrificar la eficiencia por una especificidad más alta, minimizando así los falsos positivos.
Algunos factores que controlan estos parámetros de la PCR son:
- Temperatura
- Tiempo de extensión
- Concentración de ADN molde
- Propiedades termodinámicas de la secuencia blanco y los primers
- Tamaño del amplicón
- Cationes divalentes (Mg+2 y Mn+2) y detergentes.
Criterios para el diseño
Si queremos enfocarnos en dominar la especificidad, hay otro factor que es importante considerar la sensibilidad
- Un primer no sensible no reconoce la secuencia de ADN blanco.
- Un primer no específico puede detectar y unirse a secuencias adicionales en el genoma, distintas de la secuencia de interés.
Longitud del primer
- Estadísticamente podemos asumir que las bases nitrogenadas:
- Ocurren independientemente una de otra.
- Cada composición de las bases puede distribuirse de manera idéntica.
- La probabilidad de que ocurra cualquiera de las 4 bases disponibles A, C, T, G es $\mathrm{p}{A}$, $\mathrm{p}{C}$, $\mathrm{p}{G}$, $\mathrm{p}{T}$ entonces podemos utilizar la siguiente expresión:
$$\mathrm{p}{A}=\mathrm{p}{C}=\mathrm{p}{G}=\mathrm{p}{T}=\frac{1}{4}=0.25$$
Ahora podemos calcular la frecuencia en la que podríamos encontrar una secuencia de 15 nucleótidos igual en un genoma con tamaño de $3.3\ast{10}^{9}$ pares de bases.
$$\left(\frac{1}{4}\right)^{15}\left(3.3\ast{10}^{9}\right)=3.07$$
Esto quiere decir que con un genoma de $3.3\ast{10}^{9}$ pb una secuencia de 15 nt podríamos encontrarla 3 veces a lo largo del genoma.
Es por esto que la especificidad está controlada por el tamaño del primer, a medida que aumenta su tamaño es menos probable la frecuencia para encontrarlo en el genoma.
- Un tamaño óptimo para la especificidad de un primer está entre 18 y 24 nt
- Por cada nucleótido adicional el primer se vuelve 4 veces más específico.
- El numero superior en el tamaño, es menos crítico pero puede afectar a la eficiencia de la reacción.
- Por razones de entropía, mientras más corto sea el primer, podrá alinearse más rápido al ADN molde para poder formar un duplex estable.


Primers con secuencias más largas (28-35 nt)
Sirven para amplificar secuencias con un alto grado de heterogeneidad
- Para clonación de genes homólogos de diferentes especies o isoformas de proteínas dentro de una familia de proteína de una especie en particular.
- Amplificación de secuencias virales tales como el VIH-1 donde no se espera la posibilidad de encontrar un juego de primers con la complementariedad perfecta para todas las secuencias blanco. También se necesitan cuando se añaden sitios de información extra en los primers para el extremo 5'
- Sitio de unión a T7 ARN pol
- Sitios para el reconocimiento de enzimas de restricción (añaden 6 nt extra con 2 o 3 bases no específicas a la secuencia blanco)
El tamnaño debe variar por no menos de 3 pb entre los primers forward y reverse para que puedan unirse a sus regiones en el genoma al mismo tiempo.
Estabilidad de los primers (Parámetros termodinámicos)
Propiedades de la desnaturalización del ADN
- Cuando se calienta el ADN a ${80}^\circ$C, su absorbancia UV incrementa 30-40%.
- Con el calentamiento las fuerzas no covalentes que mantienen unidas las dos cadenas del ADN se rompen.
- Cuando se vence la fuerza de union, las dos cadenas, se rompe la doble hélice
- Pueden volver a formar la doble hélice si se enfria lentamente el recipiente que las contiene
Temperatura de fusión (Tm)
Conforme las hebras se separan, se incrementa la absorbancia a 260 nm debido a que los pares de bases apilados absorben menos luz.
Este incremento de absorbancia es llamado hipercromicidad.

El término Tm representa la temperatura a la cual las cadenas de ADN están ½ (50%) desnaturalizadas que se denomina como temperatura de fusion (melting) Tm. En otras palabras es la temperatura a la cual se ha perdido la mitad de la estructura helicoidal por la desnaturalización por calor.

Relación entre la Tm y %GC
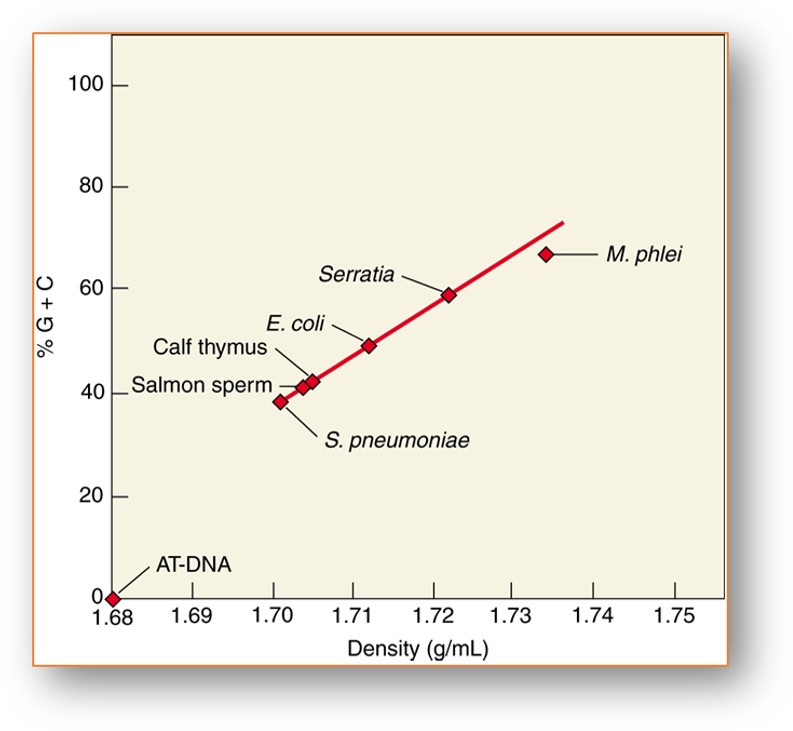
El contenido de GC del ADN tiene un efecto significativo en la Tm a mayor %GC se elevará la Tm.

En la desnaturalización, los puentes de hidrógeno entre los peres de bases complementarios se reemplazan por enlaces de hidrógeno energéticamente equivalentes entre los nucleotidos y el agua

Además del calor, el ADN puede ser desnaturalizado por:
- Solventes Orgánicos.
- pH alto.
- Alta concentración de sales.
El %GC también afecta directamente densidad del ADN es una relación lineal directa
Renaturalización del ADN
Después de que se separa la doble hélice de ADN, bajo las condiciones adecuadas ambas cadenas pueden unirse de nuevo a este proceso es llamado alineamiento o renaturalización. Los tres factores más importantes:
- Temperatura – major por debajo de 25 C abajo de la Tm.
- Concentración de ADN – dentro de los límites a una concentración más alta higher concentration tendrá más probabilidad de que las 2 cadenas complementarias se encuentren la una a la otra.
- Tiempo de renaturalización Time – conforme incrementa el tiempo, habrá mayor alineamiento.
Cálculo de Tm en el primer
Los primers forward y reverse deben tener un valor similar de Tm, de lo contrario la amplificación del ADN será menos eficiente o no funcionará
En la siguiente tabla se muestran los distintos métodos para ello
| Método | Referencias |
|---|---|
| Ecuación de Marmur y Doty | Marmur J and Doty P (1962) J Mol Biol 5:109 y Chester N, Marshak DR. (1993) Anal Biochem;209: 284 |
| Fórmula de Wallace | Wallace RB et al. (1979) Nucleic Acids Res 6:3543 |
| Método de ajuste de sales | Schildkraut C. (1965) Biopolymers.;3(2):195 yBreslauer KJ, et al. (1986) PNAS. 83(11):3746 |
| Método de los vecinos cercanos | SantaLucia J Jr (1998) PNAS. 95(4):1460-5. |
Nota: El valor de Tm depende de varios factores como el tamaño del del ADN, la composición de la secuencia, el ambiente iónico, pH, etc.
Ecuación de Wallace
Asume que la PCR se lleva a cabo en condiciones estándar
- [Primer] = 50mM
- [Na] = 50mM
- pH = 7.0
$$T_{m}=\left[{4}^\circ{C}\times\left(\text{Numero de }G+C\right)\right]+\left[{2}^\circ{C}\times\left(\text{Numero de }A+T\right)\right]$$
Ejemplo:
5’-ACAGATAGTGGGGATCCG-3’
Número de:
- G + C = 10
- A + T = 8
$$T_{m}=\left[{4}^\circ{C}\times\left(10\right)\right]+\left[{2}^\circ{C}\times\left(8\right)\right]={56}^{\circ}{C}$$
Ecuación de Marmur y Doty con la corrección de Chester y Marshak
Marmur y Doty establecieron una formula para correlacionar el contenido de GC (%GC) a la Tm de dúplex largos de ADN a una concentración iónica dada. Chester y Marshak posteriormente añadieron un término para tomar en cuenta la longitud de la cadena de ADN
Utiliza las condiciones estándar de reacción de PCR.
$$𝑇_𝑚=69.4+\frac{41\times\left[\left(\text{número de }G+C \right)-16.4\right]}{\text{número de }A+T+G+C}$$
Ejemplo:
5’-ACAGATAGTGGGGATCCG-3’
Número de:
- G + C = 10
- A + T + G + C = 18
$$𝑇_𝑚=69.4+\frac{41\times\left[10-16.4\right]}{18}=\frac{2467}{45}={54.82}^\circ{C}$$
Método de ajuste de sales
Asume que:
- [primers] = 50mM
- pH = 7.0
$$𝑇_𝑚=100.5+\left(\frac{41\times\left(\text{número de }G+C\right)}{\text{número de }A+T+G+C}\right)-\left( \frac{820}{\text{número de }A+T+G+C} \right)+\left(16.6\times\log_{10}\left[Na^+\right]\right)$$
Ejemplo:
5’-ACAGATAGTGGGGATCCG-3’
Número de:
- G + C = 10
- A + T + G + C = 18 [Na] = 50 mM = 0.05 M
$$𝑇_𝑚=100.5+\left(\frac{41\times10}{18}\right)-\left( \frac{820}{18} \right)+\left(16.6\times\log_{10}\left[0.05\right]\right)={56.13}^\circ{C}$$
Método de vecinos cercanos (Nearest-Neighbor)
Asume que:
- La estabilidad del dúplex de ADN depende de bases vecinas más cercanas (apilamiento)
- Toma en cuenta los parámetros termodinámicos de la interacción en el dúplex de ADN
- Entalpía
- Entropía
- Energía Libre de Gibbs
Entalpía
- Se refiere a la cantidad de energía calorífica liberada o absorbida por el primer
- $\Delta{H}$ mide el cambio de la entalpía
- $\Delta{H}_{\left[primer\right]}$ = La suma del valor de 𝛥𝐻 de cada par de nucleótidos que son vecinos cercanos en el primer
Valor de $\Delta{H}$ para cada par de vecinos cercanos

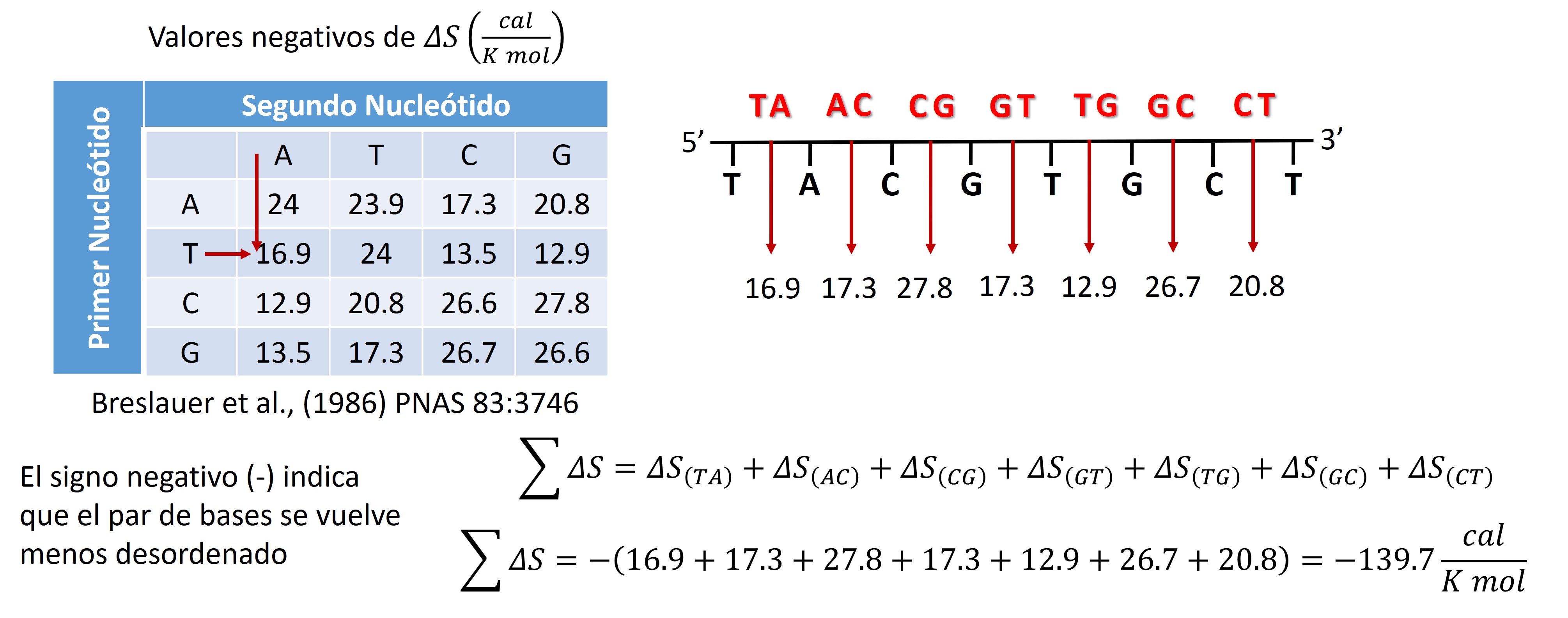
Entropía
Se refiere a La cantidad de desorden que exhibe un primer
Cuando se forma el dúplex ADN-primer, la entropía del primer y del ADN de una sola cadena disminuye, dado a que pierden su grado de libertad de movimiento cuando se unen el uno al otro. Cambian de una estado desordenado a uno ordenado.
$\Delta{S}$ mide el cambio de entropía
$\Delta{S}_{\left[primer\right]}$ = La suma del valor de $\Delta{S}$ de cada par de dinucleótidos en el primer que son vecinos cercanos
Valor de $\Delta{S}$ para cada par de vecinos cercanos
Energía Libre de Gibbs ($\Delta{G}$)
Se refiere a la energía requerida para romper una estructura secundaria no deseada en el primer, esto ocurre cuando el primer se alinea a sí mismo (harpin) o al otro primer (primer dimer).
Mientras más negativo sea el valor de $\Delta{G}$ más estable será la estructura secundaria.
El valor máximo que podemos permitir es de -9 $\frac{Kcal}{mol}$, valores más negativos requerirán de una alta energía para romper la estructura secundaria no deseada.

Cálculo de la $\Delta{G}$ del primer

Modelo Nearest-Neighbor


Ecuación Nearest-Neighbor

Estabilidad Interna
El Perfil de estabilidad se calcula con los valores de Nearest-neighbour. Usualmente se grafica el valor de ΔG para todos los nucleótidos de los primers.
Para minimizar los sitios de unión no específica (false priming), la estabilidad del extremo 5’ debe ser más alta que la estabilidad del extremo 3’

Estabilidad 5’ y estabilidad 3’
La elongación del primer comienza en el extremo 3’, por lo tanto conforme el extremo 3’ se hibrida al ADN molde de manera estable la elongación comienza.
El extremo 5’ juega un papel menos importante
Esta característica produce un problema - si el extremo 3’ del primer tiene más de 3 C/G, puede unirse de manera casi estable a cualquier sitio donde existan tres base complementarias G/C
Situación ideal:
- Extremo 5’ estable + un extremo 3’ menos estable, esto elimina los sitios de unión no específicos debido al alineamiento de la mitad del primer del extremo 3’. Se prefiere que el extremo 5’ tenga una o dos bases G/C (GC clamp) y que el extremo 3’ no tenga más de una base G/C

Temperatura de Alineamiento (Ta)
Es la temperatura a la cual los primers se alinean con el ADN
Puede ser calculada a partir de la Tm
$$T_{alineamiento}=T_{m\text{ primer}}-{4}^{\circ}C=0.3{\times}T_{m\text{ primer}}+0.7{\times}T_{m\text{ producto}}-14.9$$
Para asegurarse que los primers y al ADN molde se alinean antes que las dos hebras del templado se unan de nuevo se requiere que:
$$T_{m\text{ producto}}-T_{alineamiento}\ge{30}^{\circ}C$$
Determinación empírica de la Ta
Para determinar experimentalmente la temperatura de alineamiento, se debe hacer una PCR a varias temperaturas comenzando por ${5}^\circ{C}$ por debajo del valor mínimo de la Tm calculado para el par de primers.
$$T_{alineamiento}=\text{valor mínimo de }T_{m\text{ primer (forward o reverse)}}-{5}^\circ{C}$$

Si la electroforesis resulta en bandas inespecíficas se debe incrementar el punto de partida para la determinación empírica de la Ta por incrementos de $1$ a ${2}^\circ{C}$ , hasta que las bandas no específicas desaparezcan del gel como de ve en la sigueinte figura a partir del carril 9.

Astringencia en el alineamiento de primers
La astringencia determina la especificidad del producto no amplificado, el factor más significativo es la Ta.
- Una Ta muy baja el alineamiento es menos astringente y el primer se puede unir a otros sitios no específicos.
- Una Ta muy alta da mayor astringencia al alineamiento, pero el primer puede no unirse a la secuencia blanco.

Otros factores que pueden influir son:
- %GC: los pares de GC son más astringentes que los de AT.
- Concentración de sales y el buffer utilizado.
Para que los primers forward y reverse se unan al mismo al ADN tiempo durante el paso de alineamiento la Tm de ambos debe ser:
$$\Delta{T}_{m}\ge{5}^\circ{C}$$
Composición de las bases
La composición de bases afecta la especificidad de hibridación, la temperatura de Fusión/Alineamiento y la estabilidad interna
Se prefiere una composición aleatoria. Debemos evitar regiones largas con (A + T) y (G + C)
Usualmente un contenido promedio de 50% de (G + C) nos da una temperatura de fusión/alineamiento correcta.
Estructura interna
Si los primers pueden alinearse a ellos mismos formando estructuras secundarias (horquilla, autodímeros) o entre ellos (dímeros) en lugar de unirse al templado de ADN, habrá un decremento considerable de la eficiencia de la PCR. Esto debe ser evitado

Estas estructuras no son perjudiciales cuando la temperatura de alineamiento no permite que se formen. Por ejemplo algunos dímeros o harpins se forman a ${30}^\circ{C}$ durante un ciclo de PCR, pero la temperatura del ciclo no baja de ${60}^\circ{C}$ de esta forma esta temperatura no permite que se formen este tipo de estructuras, también hay que tener en cuenta el valor de $\Delta G$ para cada una de estas estructuras deben evitarse valores más negativos a: -9 $\frac{Kcal}{mol}$.
Los valores aceptables de $\Delta G$ para estas estructuras secundarias son:
- Harpins -2 $\frac{Kcal}{mol}$
- Dímeros internos -6 $\frac{Kcal}{mol}$
- Complementariedad Auto/cruzada extremo 3’-5 $\frac{Kcal}{mol}$
Tamaño del producto y colocación dentro de la secuencia blanco
El tamaño del producto afecta la eficiencia de la PCR
- Generalmente se amplifican productos de 150 – 1000 pb.
- Para la detección de la expresión génica, se diseñan oligos que flanquean los exones.
Se debe evitar regiones con corridas largas o repetidos.
Hay que revisar si el primer no tiene homología cruzada es decir sitios no deseados donde pueda unirse específicamente con el ADN molde.
Y también verificar si el producto de amplificación posee una estructura secundaria, y la estabilidad de ésta.
Bibliografía
- Álvarez-Fernández, R. (2013). Chapter One - Explanatory Chapter: PCR Primer Design. InLorsch, J. (Eds.), Methods in Enzymology. (pp. 1–21). Academic Press. https://doi.org/10.1016/B978-0-12-418687-3.00001-X
- Appling, D., Anthony-Cahill, S. & Mathews, C. (2016). Chapter 4: Nucleic Acids. In Biochemistry Concepts and Connections. (1st, pp. 104–139). Pearson Education.
- Breslauer, K., Frank, R., Blöcker, H. & Marky, L. (1986). Predicting DNA duplex stability from the base sequence.. Proceedings of the National Academy of Sciences, 83(11). 3746–3750. https://doi.org/10.1073/pnas.83.11.3746
- Chester, N. & Marshak, D. (1993). Dimethyl Sulfoxide-Mediated Primer Tm Reduction: A Method for Analyzing the Role of Renaturation Temperature in the Polymerase Chain Reaction. Analytical Biochemistry, 209(2). 284–290. https://doi.org/10.1006/abio.1993.1121
- Dieffenbach, C., Lowe, T. & Dveksler, G. (1993). General concepts for PCR primer design.. Genome Research, 3(3). S30–S37. Retrieved from https://genome.cshlp.org/content/3/3/S30
- Dieffenbach, C., Lowe, T. & Dveksler, G. (1993). General concepts for PCR primer design. PCR methods and applications, 3(3). S30–37. https://doi.org/10.1101/gr.3.3.s30
- Aoyagi, K. (2001). PCR. In Molecular biology problem solver. (pp. 291–329). John Wiley & Sons, Ltd. https://doi.org/https://doi.org/10.1002/0471223905.ch11
- Marmur, J. & Doty, P. (1962). Determination of the base composition of deoxyribonucleic acid from its thermal denaturation temperature. Journal of Molecular Biology, 5(1). 109–118. https://doi.org/https://doi.org/10.1016/S0022-2836(62)80066-7
- Rychlik, W. (2000). Primer selection and design for polymerase chain reaction. InRapley, R. (Eds.), The nucleic acid protocols handbook. (pp. 581–588). Humana Press. https://doi.org/10.1385/1-59259-038-1:581
- Rychlik, W. (1993). Selection of primers for polymerase chain reaction. Methods in Molecular Biology (Clifton, N.J.), 15. 31–40. https://doi.org/10.1385/0-89603-244-2:31
- SantaLucia, J. (1998). A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proceedings of the National Academy of Sciences, 95(4). 1460–1465. https://doi.org/10.1073/pnas.95.4.1460
- Sariera, S.(2020). Retrieved from https://easyexperiment.teachable.com/p/pcr-primer-design
- Schildkraut, C. & Lifson, S. (1965). Dependence of the melting temperature of DNA on salt concentration. Biopolymers, 3(2). 195–208. https://doi.org/10.1002/bip.360030207
- Wallace, R., Shaffer, J., Murphy, R., Bonner, J., Hirose, T. & Itakura, K. (1979). Hybridization of synthetic oligodeoxyribonucleotides to phi chi 174 DNA: the effect of single base pair mismatch.. Nucleic Acids Research, 6(11). 3543–3557. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC327955/
- Wikimedia Commons(2013). Retrieved from https://commons.wikimedia.org/wiki/File:Primers_RevComp.svg
{kind=link}