Hola soy Felipe
Este blog tiene archivados algunos archivos históricos de bioinformática, donde las herramientas mencionadas ya no estan disponibles actualmente
El contenido de este post puede ser abierto de forma interactiva en Google Colab Rychlik (1993) publicó una guía sobre la selección de primers para la Reacción en Cadena de la polimerasa (PCR) donde menciona una gráfica de estabilidad interna para los oligos, la cual puede obtenerse utilizando Oligo 7. Esta gráfica puede ayudarnos a evitar el mispriming (sitios falsos de unión a la secuencia blanco) a partir del valor de $\Delta G$ de los pentámeros del oligo....
El término bioinformática conceptualiza la biología en términos de moléculas (proteínas, ácidos nucléicos) y la aplicación de técnicas informáticas para entender y organizar este tipo de información biológica. Así que su meta principal tiene que ver con poder encontrar información guardada dentro de los organizamos vivos y depende de conceptos y aplicaciones de la biología, las ciencias de la computación y el análisis de datos. La bioinformática es predictiva Para poder encontrar el sentido de los datos biológicos debemos hacer predicciones acerca de lo que ya conocemos, es decir obtener conocimiento biológico por comparación con lo que se conoce....
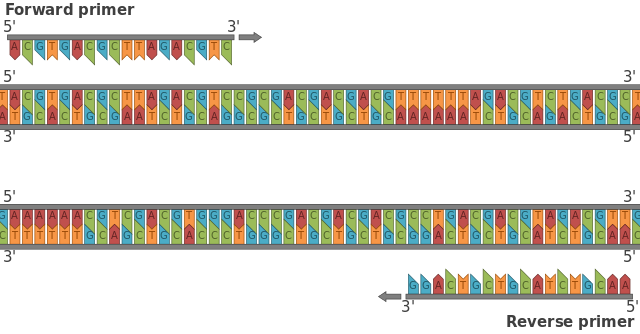
¿Qué es un primer? Los primers son secuencias cortas de moléculas de ácidos nucléicos (entre 18 a 24 pares de bases) que son utilizados para la amplificación de un gen o un fragmento de ADN de interés, mediante la Reacción en Cadena de la Polimerasa (PCR). De tal forma que los cebadores o primers, son uno de los principales ingredientes para una reacción de PCR, y de ellos depende la especificidad, porque al unirse complementariamente a las dos cadenas de ADN de la secuencia molde, fijan por así decirlo las coordenadas donde se llevará acabo la reacción....
La transferencia de proteínas o ácidos nucléicos a una membrana inmovilizante, es referida como “blotting”, la transferencia puede ser realizada por difusión molecular, por un flujo de buffer inducido por la succión (microfiltración) o acción capilar, el método utilizado de manera más frecuente es la electrotransferencia. La difusión es un método simple pero muy ineficiente para aplicar a la mayoría de las transferencias, exepto para las transferencias de geles de agarosa....
El western blott o inmunoblott es un inmunoensayo en el cuál anticuerpos específicos son utilizados como sondas marcadas. Esta técnica permite comparar la abundancia de las proteínas separadas por electroforesis en gel, utilizando carriles distintos para cada una de las muestras. La técnica puede dividirse en 5 pasos Electroforesis Blotting Inmunodetección Imagen Análisis (Cuantificación) Electroforesis Cualquier molécula cargada en solución, migrará en un campo eléctrico aplicado, un fenómeno conocido como electroforesis....